台灣GMP認證
聯絡我們-親切顧問師團
隊改變您對認證想法
台灣總部:
台灣台中市沙鹿區中山路19巷13號
No.13, Ln. 19, Zhongshan Rd., Shalu Dist., Taichung City 433,
Taiwan (R.O.C.)
服務專線:04-26153935
CE專線:04-23806957
傳真:04-26154055
info@avtce.com.tw

GMP證書範例:


台灣醫療器材優良製造規範認GMP
依據醫療器材管理辦法的規定,將醫療器材依據風險程度,分成第一等級(低風險性)、第二等級(中風險性)、第三等級(高風險性),除醫療器材管理辦法附件二未滅菌者以及不具量測功能的醫療器材適用精要模式之外,其餘所有醫療器材都須申請符合醫療器材優良製造規範之標準模式;而未滅菌者及不具量測功能的醫療器材,不論國產醫療器材製造業者或輸入醫療器材之販賣業者應符合藥物優良製造準則第三章精要模式之規定,得無須提出符合性申請,相關品質系統文件留廠備查,並配合衛生福利部食品藥物管理署執行不定期稽核時,提出相關文件供查核。
「藥物製造業者檢查辦法」規定衛生機關對於藥物(含醫療器材)製造業者之檢查有四
種:
- 新設、遷移、擴建、復業或增加原料藥、劑型、加工項目、品項之檢查
- 後續追蹤管理之檢查
- 區域例行性檢查
- 其他檢查
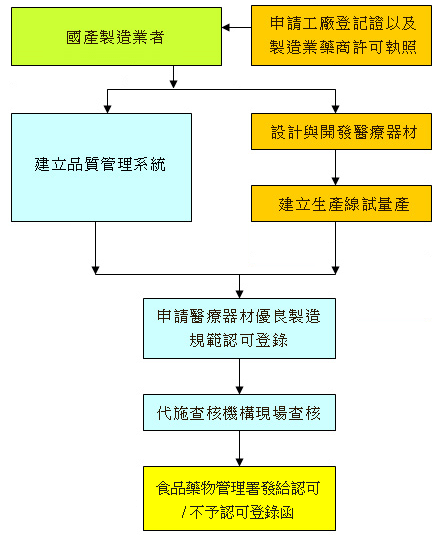
上述第一項的檢查,國產醫療器材製造業者須完成以下兩階段的檢查作業,經檢查符合規定者,由衛生福利部食品藥物管理署就檢查合格之項目,核發符合醫療器材優良製造規範之證明文件:
- 其硬體設備及衛生條件,應符合藥物製造工廠設廠標準第二編及工廠管理輔導法之規定,並由工業主管機關及直轄市或縣(市)衛生主管機關檢查。符合規定者,由直轄市或縣(市)工業主管機關依申請核發工廠登記證明文件或核准變更登記,並由直轄市或縣(市)衛生主管機關依申請核發製造業藥商許可執照或核准變更登記。
- 其軟體設備及衛生條件,應符合藥物優良製造準則第三編「醫療器材優良製造規範」之規定,如經檢查符合規定者,由中央衛生主管機關就檢查合格之項目,核發符合醫療器材優良製造規範之證明文件。
輸入醫療器材製造工廠檢查方式以審查品質系統文件(Quality System Documentation,QSD)為主,輸入藥商也可選擇申請海外查廠。
醫療器材優良製造規範
國產及輸入業者須確認原廠的品質管理系統是否依據前行政院衛生署食品藥物管理局(現今為衛生福利部食品藥物管理署)於102年3月11日公告之藥物優良製造準則第三編「醫療器材優良製造規範」及ISO 13485:2003所建立。
| 醫療器材優良製造規範要求項目 | ISO 13485:2003 |
|
第一節 |
4 Quality management system |
|---|---|
| 第63條 | 4.1 General requirements |
| 第64條 | 4.2.1 General, Documentation requirements |
| 第65條 | 4.2.2 Quality manual |
| 第66條 | 4.2.3 Control of documents |
| 第67條 | 4.2.4 Control of records |
| 第二節 | 5 Management responsibility |
| 第68條 | 5.1 Management commitment |
| 第69條 | 5.2 Customer focus |
| 第70條 | 5.3 Quality policy |
| 第71條 | 5.4.1 Quality objectives |
| 第72條 | 5.4.2 Quality management system planning |
| 第73條 | 5.5.1 Responsibility and authority |
| 第74條 | 5.5.2 Management representative |
| 第75條 | 5.5.3 Internal communication |
| 第76條 | 5.6.1 General, Management review |
| 第77條 | 5.6.2 Review input |
| 第78條 | 5.6.3 Review output |
| 第三節 | 6 Resource management |
| 第79條 | 6.1 Provision of resources |
| 第80條 | 6.2.1 General, Human resources |
| 第81條 | 6.2.2 Competence, awareness and training |
| 第82條 | 6.3 Infrastructure |
| 第83條 | 6.4 Work environment |
| 第四節 | 7 Product realization |
| 第84條 | 7.1 Planning of product realization |
| 第85條 | 7.2.1 Determination of requirements related to the product |
| 第86/87條 | 7.2.2 Review of requirements related to the product |
| 第88條 |
7.2.3 Customer communication |
| 第89條 | 7.3.1 Design and development planning |
| 第90條 | 7.3.2 Design and development inputs |
| 第91條 | 7.3.3 Design and development outputs |
| 第92條 | 7.3.4 Design and development review |
| 第93條 | 7.3.5 Design and development verification |
| 第94條 | 7.3.6 Design and development validation |
| 第95條 | 7.3.7 Control of design and development changes |
| 第96條 | 7.4.1 Purchasing process |
| 第97條 | 7.4.2 Purchasing information |
| 第98條 | 7.4.3 Verification of purchased product |
| 第99條 | 7.5.1.1 General requirements, Control of production and service |
| 第100條 | 7.5.1.2.1 Cleanliness of product and contamination control |
| 第101條 | 7.5.1.2.2 Installation activities |
| 第102條 | 7.5.1.2.3 Servicing activities |
| 第103條 | 7.5.1.3 Particular requirements for sterile medical devices |
| 第104條 | 7.5.2.1 General requirements,
Validation of processes for production and service provision |
| 第105條 | 7.5.2.2 Particular requirements for sterile medical devices |
| 第106條 | 7.5.3.1 Identification |
| 第107條 | 7.5.3.2.1 General, Traceability |
| 第108條 | 7.5.3.2.2 Particular requirements for active implantable medical devices and implantable medicaldevices |
| 第109條 | 7.5.3.3 Status identification |
| 第110條 | 7.5.4 Customer property |
| 第111條 | 7.5.5 Preservation of product |
| 第112條 | 7.6 Control of monitoring and measuring devices |
| 第113條 | 8.1 General |
| 第114條 | 8.2.1 Feedback |
| 第115條 | 8.2.2 Internal audit |
| 第116條 | 8.2.3 Monitoring and measurement of processes |
| 第117條 | 8.2.4.1 General requirements, Monitoring and measurement of product |
| 第118條 | 8.2.4.2 Particular requirement for
active implantable medical devices and implantable medical devices |
| 第119條 | 8.3 Control of nonconforming product |
| 第120條 | 8.4 Analysis of data |
| 第121條 | 8.5.1 General, Improvement |
| 第122條 | 8.5.2 Corrective action |
| 第123條 | 8.5.3 Preventive action |
國產醫療器材製造業者
根據藥事法第十八條規定,所謂醫療器材製造業者,係指製造、裝配醫療器材,與其產品之批發、輸出及自用原料輸入之業者,此外,藥事法對藥物製造工廠之相關規定亦明訂於第五十七條(藥物之製造與設廠標準)與第五十八條「藥物工廠,非經中央衛生主管機關核准,不得委託他廠製造或接受委託製造藥物」。
委託製造
醫療器材製造業者若委託或接受委託製造,必須依據「藥物委託製造及檢驗作業準則」的規定辦理。所謂委託製造,係指將藥物在製程中之任一階段或連續階段或全程委託他廠製造(第三條)。委託製造藥物,依法應事先申請中央衛生主管機關核准,而實務上,製造廠可先取得醫療器材優良製造規範認可登錄函,並於查驗登記時檢具委託者及受託者雙方出具之委託製造證明文件,同時申請。另申請藥物委託製造,限持有藥物許可證或申請藥物查驗登記之藥商(第五條)。經核准委託製造之藥物,除法律另有規定外,其產品責任由委託者負責(第六條)。
最後,經核准委託製造之藥物,其標籤及包裝,除應符合本法及有關法令規定外,並應刊載受託廠及委託者之名稱、地址。但受託廠之名稱、地址,經中央衛生主管機關核准者,得以刊載其所在國別替代之(第十條)。
嚴重不良反應通報與藥物回收
除醫療器材優良製造規範之外,國內外製造業者須依據「藥物回收作業實施要點」以及「嚴重藥物不良反應通報辦法」建立適當的標準作業程序,以供執行。
|
危害等級 |
定義 |
回收規定 |
|---|---|---|
|
第一級危害 |
係指藥物有下列情形之一者:
|
中央、直轄市或縣(市)衛生主管機關應命藥物製造或輸入之業者依規定期限內回收市售品。 |
|
第二級危害 |
係指藥物有下列情形之一者:
|
中央、直轄市或縣(市)衛生主管機關應命藥物製造或輸入之業者依規定期限內回收市售品。 |
|
第三級危害 |
係指藥物有損害使用者安全、健康或其他權益之虞,而非屬第一級或第二級危害者。 | 中央、直轄市或縣(市)衛生主管機關得建議藥物製造或輸入之業者自衛生主管機關通知之日起六個月內收回市售品。 |
「嚴重藥物不良反應通報辦法」規定因藥物所引起之嚴重藥物不良反應發生時,醫療機構、藥局、藥商應依本辦法填具通報書,連同相關資料,向中央衛生主管機關或其委託機構通報。
|
嚴重藥物不良反應之定義 |
藥商通報要求 |
|
|
體外診斷試劑
由於體外診斷醫療器材的產品特性與製造方法有其特殊性,前衛生署(現今衛福部)特別制定並公佈「體外診斷試劑優良製造規範指導手冊」,可供醫療器材製造業者參考,其內容如下表:
|
貳、品質系統之要求 |
|
|
四、品質系統 |
十七、供水系統 |
|
五、產品規格 |
十八、加熱、通風與空調系統 |
|
六、製程規格 |
十九、污染/交叉污染 |
|
七、製程確效 |
二十、電腦系統 |
|
八、製程及製程管制 |
|
|
九、環境管制 |
|
|
十、作業人員之穿著 |
|
|
十一、清潔與衛生 |
|
|
十二、組件 |
|
|
十三、成品檢驗與測試 |
|
|
十四、安定性試驗及有效期限 |
|
|
十五、怨訴與失效調查 |
|
|
十六、品質趨勢分析 |
|
海外製造廠
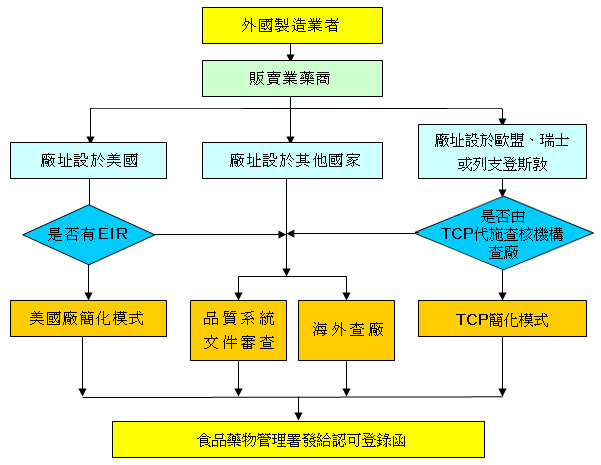
- 6-1品質系統文件審查
- 該輸入醫療器材國外製造業者之品質系統文件。申請人得先檢附品質手冊與相關程序書及文件總覽表。但必要時,申請人應依衛福部之通知,補送其他品質系統文件或資料。
- 與醫療器材優良製造規範同等效力之符合性驗證合格登錄證書。
- 該輸入醫療器材國外製造業者之全廠配置圖、各類產品製造作業區域、主要設備、產品製造流程;必要時,並應標示作業人員與物料搬運之通路。
產地為美國之製造業者,得以美國最高衛生主管機關出具之製售證明,並其內容載明該製造業者係符合美國之醫療器材優良製造規範(Current Good Manufacturing Practice)者,替代「與醫療器材優良製造規範同等效力之符合性驗證合格登錄證書。」
- 6-2 美國廠簡化模式
產地為美國、美屬波多黎各或關島之製造業者,於中美醫療器材技術合作換文有效期間內,得以美國最高衛生主管機關出具之查廠報告書與製售證明及與醫療器材優良製造規範同等效力之符合性證書(ISO 13485),共同替代前6-1所述資料。
- 6-3歐盟簡化模式
歐盟技術合作方案(或與我國簽訂合作換文之國家/地區)簡化模式(僅適用廠址位於歐盟地區之製造廠,且以參與「台歐查廠報告技術合作方案」之歐盟代施查核機構為限,或與我國簽訂合作換文之國家/地區亦得適用),得以歐盟(或與我國簽訂合作換文之國家/地區)代施查核機構出具之最近一次查廠報告,連同該歐盟會員國最高衛生主管機關出具之製售證明及該歐盟受託查核機構出具與醫療器材優良製造規範同等效力之符合性證書(如ISO 13485),共同替代前6-1 所述資料。
- 6-4 海外查廠
依據藥物製造業者檢查辦法第7條第一項檢查如有實施國外檢查之必要者,申請人應向衛福部繳納費用及國外製造業者之品質手冊,並與國外製造業者配合檢查要求,備齊相關資料。
出處及延伸閱讀:
- 「藥事法」,中華民國一百零二年五月八日總統華總一義字第 10200082721 號令修正公布第 13 條條文
- 「藥物優良製造準則」,中華民國一百零二年三月十一日行政院衛生署署授食字第 1021100245號令訂定發布全文 146 條;並自發布日施行
- 「藥物製造工廠設廠標準」,中華民國一百零二年三月十一日行政院衛生署署授食字第 1011103264 號令、經濟部經工字第 10204600840 號令會銜修正發布全文 34條;並自發布日施行
- 「藥物製造業者檢查辦法」,中華民國一百年七月六日行政院衛生署署授食字第 1001100780 號令、經濟部經工字第 10004604010 號令會銜修正發布第 4、6、8~10 條條文
- 「消費者保護法」,中華民國100年12月16日行政院院臺規字第 1000109431 號公告修正發布。
附圖一:國產製造業者取得醫療器材優良製造規範認可登錄流程
附圖二:國外製造業者取得醫療器材優良製造規範認可登錄流程
幫助服務區
若您需要任何幫助,可以通過幾下方式與我司取得聯繫,我們會儘快為提供準確、周到的服務,感謝您的訪問,建議收藏此頁內容。
服務專線:04-26153935 CE專線:04-23806957 傳真:04-26154055
mail: info@avtce.com.tw
台灣歐測驗證科技股份有限公司(EU CERTIFICATE)能為您提供服務
針對歐盟機械產品CE認證、電器安規CE認證、EMC CE認證、醫療CE認證、壓力設備CE認證、建築設備CE認證、個人防護CE認證、體外醫療設備CE認證、燃燒器具CE認證..等CE諮詢服務及核發CE證書。
針對ISO體系ISO 9001 、ISO 2200、ISO 13485..各項ISO諮詢輔導驗證。
針對全球各國認證俄羅斯GOST認證、美國FCC認證、FDA認證、中國CCC認證..等諮詢輔導取證。